
Clinical trial innovations don't scale. Here's why, and how to fix it.
A Policy Agenda for Efficient, Abundant Clinical Trials

Clinical trial innovation has a scaling problem. For years, reformers have pushed innovative ideas to improve trial design and make trials more efficient. Too often, however, these ideas are presented in research papers, demonstrated in pilots, or even codified in FDA guidance documents—only to fail to become routine practice.
The contrast with other industries is stark. In other industries, new, more efficient approaches to doing business quickly replace old ones. New, cheaper service providers threaten entrenched incumbents. Innovation and competition steadily drive down costs. Trials are different. In clinical trials, the forces of innovation and competition don’t drive down costs; instead, old habits persist and bureaucracy accumulates.
If we want faster, more abundant trials, we need to understand why clinical trial innovations have failed to scale, and what policies can help.

We have an unprecedented opportunity to transform trials. We might waste it.
For decades, clinical trials – particularly in the United States – have been too slow and inefficient. They have failed to keep pace with the increasing pace of biological discovery. But today, there is an unprecedented opportunity to change that.
Several factors have come together to create this opportunity. First, there is a sense of crisis: Policymakers have noticed that the U.S. is falling behind in clinical trials and drug development, ceding leadership to countries like China. And there is growing recognition that as AI and other breakthroughs speed up drug discovery, our clinical trial system is an increasing bottleneck to scientific and medical progress.
Then, of course, there is the looming prospect of transformative technological change. In the Bay Area, where I live, a growing number of startups are entering the field, hoping to use AI to streamline and automate much of the tedious work of running clinical trials. The FDA is getting involved too through its real-time clinical trials pilot program, which is exploring the use of AI to speed up trials.
Perhaps most importantly, we have a growing movement for clinical trial abundance: an informal coalition that is pushing for better, faster trials. We’ve seen a flurry of activity in Washington DC, with excellent and innovative ideas for trial reform coming from multiple sources. Many of my favorite ideas come from my colleagues at IFP, who kick-started the clinical trials abundance project and are embracing regulatory transparency and reform. I am also excited about a new effort in Congress to improve our trial infrastructure and integrate trials into routine care.
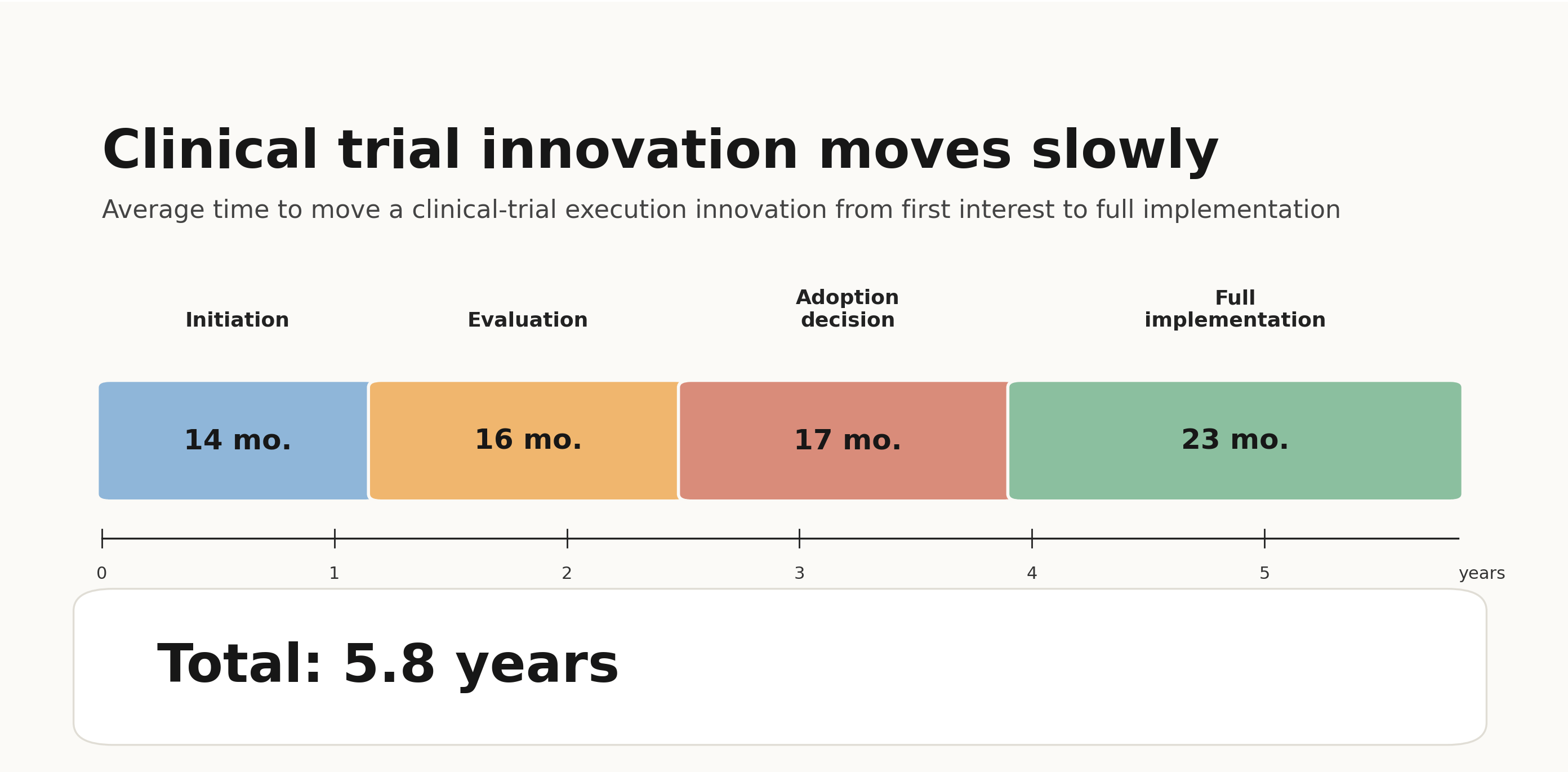
But this is not the first time we’ve reached a critical inflection point for clinical trials. And so far, we have failed to actually inflect. In 2012, the National Academy of Medicine envisioned a transformed clinical trials enterprise by 2020. Then, in the mid-2010s, with the spread of electronic health records and the passage of the 21st Century Cures Act, many anticipated a new era of digitally enabled trials. Yet most trials today continue to collect data on paper. In the early 2020s, we hoped that COVID would drive the industry towards decentralized trials, only to find that the industry retreated from decentralized trials after the pandemic ended.
Each time, the pattern was the same: we tried to reform the clinical trials system, but we never actually fixed the scaling problem. As a result, the promised reforms never materialized and the cost of trials continued to rise.
Why Clinical Trials Can’t Scale Up Innovation
Why do clinical trial innovations fail to scale? There are many reasons, but I want to highlight three major drivers.
The first is regulatory uncertainty. Drug development is an inherently risky enterprise, but too often trial innovations are not adopted because companies are uncertain about what FDA will accept. The FDA tries to provide clarity by giving sponsors — the drug companies who run the study — with early feedback and guidance, but that feedback is not sufficient to allay industry concerns. Worse yet, regulatory uncertainty tends to lead to a “compliance ratchet.” Inevitably, as companies accumulate experience they experience regulatory setbacks—they may receive negative inspection reports or get negative feedback from reviewers. To forestall potential violations and reduce regulatory risk, they impose new processes and procedures on themselves. Even if FDA issues no new regulations, this process tends to increase bureaucratic burden over time.
Regulatory uncertainty does not just limit sponsors’ own willingness to innovate; it also hinders industry-wide cooperation and trust. Or, to use the language of economists: it drastically increases transaction costs. Each drug company interprets and manages regulatory risk differently, and each feels obligated to verify—often in obtrusive and heavy-handed ways—that they are not exposing themselves to regulatory risk through their business relationships. Regulatory uncertainty leads to defensive contracting practices and intrusive and expensive monitoring and auditing between business partners. And it pushes sponsors towards narrow circles of trust that exclude new companies, new trial sites, and new technologies.
Another reason that trials industry is slow to adopt innovations is because the trials lack a reusable, shared operational infrastructure. Much of the industry still relies on fragmented, non-interoperable software and highly customized trial-by-trial workflows. In too many cases, each sponsor, site, and vendor rebuilds similar capabilities from scratch. Data must be re-entered, reconciled, and translated across systems.
The fragmentation is a result of a poorly aligned business model. Trial sites need stable workflows that work across all of the studies they conduct. But sponsors prefer that trial sites use their own software and systems to assure that their studies are being conducted correctly. The result is that sites must constantly adapt to competing sponsor demands, rather than developing their own reusable infrastructure.
Without reusable, shared operational infrastructure, it is difficult to build scalable systems to automate trials, particularly across multiple sites and sponsors. When software is used, sponsors and study sites must pay an integration tax: extra time and cost needed to integrate different pieces of software for each site, each trial, and each technology stack. This drives costs higher; in many cases, sponsors and sites forego technology and automation altogether in light of these costs.
Finally, the industry lacks coordination and shared governance to advance reforms. The trials industry is inherently fragmented: it relies on collaboration between a large and diffuse set of sponsors, sites, and vendors. But a lack of standards, coordination, and governance makes this problem worse and slows down adoption of new technologies and approaches. Each participant in the clinical trials ecosystem tends to work on their problem in isolation. While there are organizations that develop and recommend improvements to trial design and operations, these improvements are not widely adopted. There are no bodies or standards-setting organizations with the authority and ability to drive system-wide improvements.
Without common standards, shared infrastructure, and predictable regulations, each innovation must be reinvented for each sponsor, each site, and each trial.
Policies to Scale Trial Innovation
To make trials more efficient and abundant, we need a clinical trials system that can adopt and scale efficiency-improving innovations. That includes modern designs, like Bayesian, platform, decentralized, and point-of-care trials. It also includes modern approaches to trial operations, like risk-based monitoring and—at the very least—a shift away from paper-based data collection.
That’s where the proposals below come in. They target the root causes of clinical trial stagnation. These proposals aim to move us away from our current fragmented, slow, and artisanal approach to running trials to a new model in which the best designs, technologies, and operational approaches scale up and become the default, rather than the exception.
Proposal 1: The FDA should formally recognize modern, high-quality clinical trial operating practices
Anyone who studies or works on day-to-day clinical trial operations is presented with a paradox. The industry itself is deeply bureaucratic. Regulation and red tape seem to be everywhere. Yet if you read through the FDA regulations looking for the source of all of that bureaucracy, you will struggle to find it. Clinical trials are governed by a set of “good clinical practice” (GCP) guidelines and regulations that are, on their face, quite reasonable: They lay out sensible steps that drug companies and researchers should take to protect study participants and ensure that the data produced by the trials is reliable.
But something odd happens when these regulations are translated into day-to-day trial operations. Basic regulatory requirements are transformed into elaborate systems of monitoring, auditing, and procedure. That’s because the regulations are designed to provide flexibility, but they often create ambiguity as well. Reasonable people may disagree on the appropriate approach to achieving compliance with GCP, and it is difficult for sponsors to know in advance whether FDA reviewers and inspectors will deem their chosen approach acceptable. Too often, this ambiguity is resolved through “overcompliance”: a tendency for drug companies to interpret the rules in a maximalist, risk-averse way to minimize the chance of a violation.
These risk-averse interpretations can make compliance unnecessarily costly. A prominent example of this is the continued use of 100% source data verification; a time-consuming, expensive, and labor-intensive approach to verifying the accuracy of the transcription of trial data. The GCP regulations do not require 100% source data verification, as FDA has repeatedly made clear. Yet the vast majority of trials still adopt the approach, despite the fact that it consumes 25-40% of the clinical trial budget. In part, that’s because drug companies fear that FDA won’t find an alternative approach acceptable.
Risk-averse interpretations of FDA regulations do not just increase trial costs—they also limit adoption of novel and more patient-centric approaches to conducting trials. For example, for years the industry has sought to support decentralized trials which allow trial participants to use telemedicine and local health care providers instead of having to travel to distant trial sites. But ASCO has noted that regulatory ambiguity limits adoption of these trials, and that “FDA documents [related to the responsibilities of study investigators in decentralized trials] are confusing and sometimes contradictory.”
The FDA’s current set of regulatory tools are not well suited to addressing this uncertainty—particularly when it comes to the day-to-day operations of clinical trials. The FDA inspects trial sites, but these inspections focus on addressing violations after they occur; not on pre-specifying how sponsors should comply in advance. FDA’s early phase meetings, where sponsors seek feedback on proposed drug development approaches, typically focus on questions of trial design while leaving details of trial operations unaddressed. And while the FDA provides guidance to industry to address questions drug companies may have about appropriate trial practices, this guidance is typically designed to address a wide range of use cases and contexts and preserve agency discretion. It rarely provides detailed, context-specific advice about how to implement modern trial operations that drug companies and research sites can rely on.
The FDA needs new tools to increase regulatory predictability and drive adoption of modern clinical trial practices, particularly in trial operations. That’s why the FDA should create a formal Clinical Trial Practice Recognition Program. The purpose of such a program would be to recognize concrete, reusable ways of conducting clinical trials that FDA agrees are acceptable for regulatory review and inspection. Working with stakeholders, standards-development organizations, and expert consortia, the FDA would identify “recognized clinical trial practices”. These practices would codify modern, efficient approaches to trial operations, including clear specifications for how the operational approaches should be conducted and verified.
Use of an FDA-recognized clinical trial practice should come with regulatory benefits. The FDA might specify that a practice is sufficient to support review, generate reliable records, or demonstrate adherence to particular GCP expectations. And where the practice is sufficiently mature and objectively verifiable, FDA could provide assurance that use of the practice creates a presumption of acceptability for inspection or review; in this case the recognized trial practice would serve as a regulatory safe harbor.
This approach would be especially useful in areas—like source data verification and decentralized trials—where regulatory ambiguity leads to overcompliance or slows adoption of better operating models. For example, FDA could recognize practical approaches to risk-based monitoring and targeted source data verification; clarify investigator responsibilities in decentralized trials; identify acceptable documentation practices for trial master files and electronic records; or specify when digital audit trails can serve as evidence of trial conduct and oversight.
Proposal 2: The government should establish interoperability standards for clinical trial software
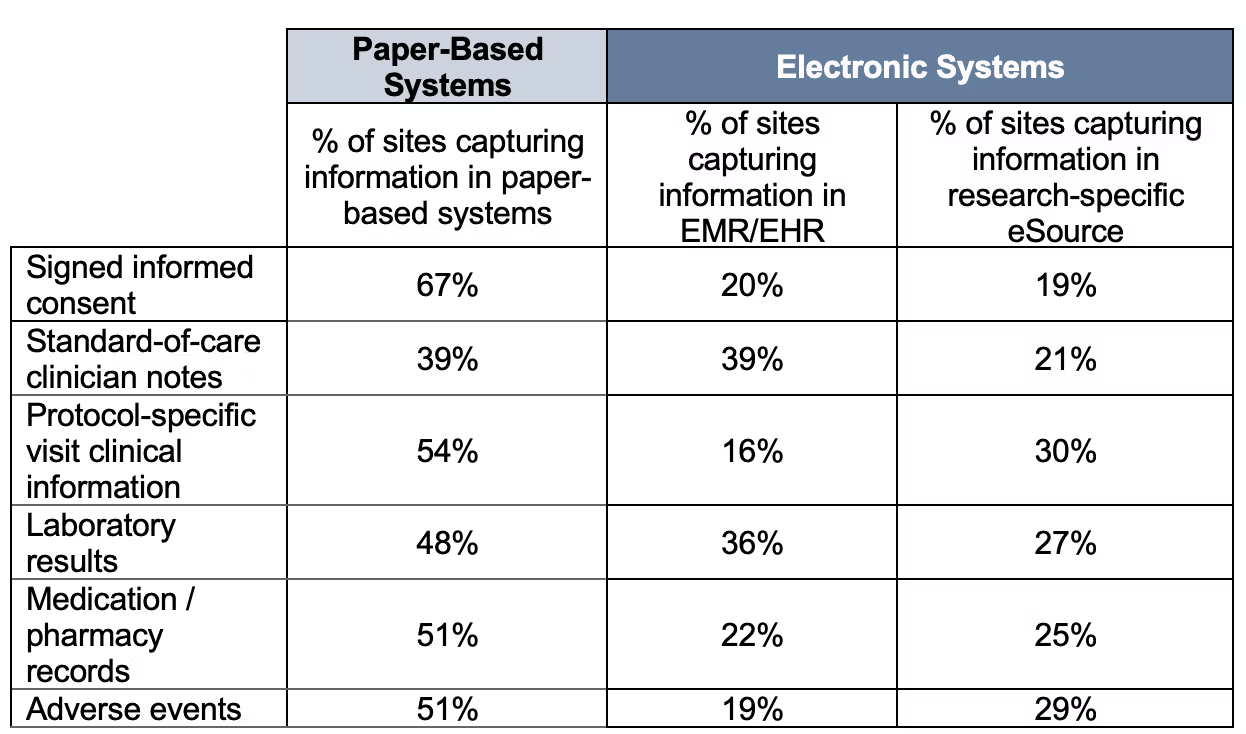
Clinical trials lag behind many other industries in their use of digital technology. At trial sites, data is still routinely collected on paper. Even when electronic systems are used, they are deeply fragmented: a single study site might use 10-20 different sponsor and vendor-specific portals to manage their trials, none of which talk to each other—and many of which don’t communicate with the study site’s electronic health records. Part of the problem is that each study sponsor often asks sites to use its own software and systems, leading to a proliferation of non-interoperable technology.
Technology fragmentation makes trials less reliable and more expensive. Trial staff must manually transcribe information across systems, slowing down operations and introducing the risk of error. Study monitors and auditors are forced to travel to research sites to conduct expensive and time-consuming audits of paper and non-interoperable digital records. Administrative tasks like billing, contracting, and documentation are also slowed, contributing to major delays when trials start up and when protocols change. These tasks take away from time that could be spent ensuring study participants are safe and adhere to the study protocol.
Fragmented technology also prevents innovative trial approaches from being adopted. Approaches like remote monitoring, real-time clinical trials, decentralized trials, adaptive trials, and seamless trials all require rapid exchange of data between systems run by sites and sponsors. These innovations are not feasible if the systems can’t talk to one another and move data seamlessly. And there is little hope of using AI to automate trials when much of the data is paper-based or locked away in inaccessible systems.
The trials industry recognizes the fragmentation problem but has been unable to solve it. One problem is that the industry’s main participants have competing incentives: trial sites want a consistent approach and workflow across every trial they run, and they want systems that integrate well with their electronic health records. Sponsors, on the other hand, want sites to use systems that integrate with their own systems. Most often, sites are forced to use different software for each sponsor they work with, driving further fragmentation. In many cases, sites resort to paper rather than contending with the integration challenges.
To fix this, we need to ensure that the different systems used by sponsors and sites can work together. We have solved this kind of challenge before in health care: Over the past decade, with support from the government, health systems have improved their ability to securely share data and exchange it between systems to support patient care. It’s time to do the same for clinical trial research.
We should establish a clinical trials technology interoperability and certification program. This program should be administered by the Office of the National Coordinator for Health Information Technology (ONC), the federal office that manages interoperability policy for health care. Under this program, ONC, collaborating closely with FDA, would set basic certification standards for trial software. Those standards would establish common ways for software to share data and provide assurance to sponsors that the software is capable of protecting data integrity and patient privacy.
An interoperability and certification program would strengthen the clinical trials ecosystem in several ways. It would speed up adoption of new and innovative software tools that support innovative trial designs. It would also strengthen competition in the clinical trial software market: shared standards would allow new and innovative technologies to quickly become a trusted part of the ecosystem, prevent incumbent software companies from building data moats and blocking access to competitors, and create a shared digital infrastructure that benefits everyone.
Proposal 3: The government should run more phase III clinical trials—and build infrastructure for them
The costs and benefits of innovative clinical trial approaches are imbalanced. When a drug company adopts a clinical trial innovation, their experience benefits the entire industry: both industry and FDA learn from the experience, and the innovation is “de-risked” for others. However, the risk is borne entirely by the first innovator. They suffer the consequences if the innovative trial approach does not produce reliable and trustworthy data or is not accepted by FDA.
Because of this risk imbalance, the industry faces a collective action problem: nobody wants to be the first (or even second) to try an innovative trial approach. They would rather see someone else do it. As a result, many innovative approaches are never attempted. For example, since 2024, FDA has been trying to recruit sponsors into a demonstration program to advance streamlined clinical trials embedded in clinical practice. To encourage them, FDA offered sponsors close collaboration and frequent feedback. Yet as of April 2026, not a single sponsor has participated in the program. (One sponsor did sign up and subsequently withdrew.)
One-off pilot projects can help de-risk new trial approaches, but they are often insufficient to change practice for three reasons: First, pilots don’t necessarily resemble industry’s own registrational trials, so they typically do not give drug companies sufficient assurance that an innovative approach will be accepted by FDA and other regulators. Second, a single pilot may be insufficient to motivate industry to adopt an innovation; drug companies would prefer to see repeated successes. And third, the innovations tested in pilots are not readily reproducible. For innovations to be adopted more widely, they must be more than just a proof-of-concept: they must generate reusable infrastructure, tools, and know-how that others can leverage. A single pilot is rarely sufficient to achieve this.
If we want to truly de-risk innovations, we need them to be regulator-ready, repeated, and reusable. The government can help: NIH should fund more large-scale “phase 3-type” trials that apply innovative approaches to reducing trial costs and increasing patient access. NIH already funds basic research and early-phase trials, but funds relatively few large-scale confirmatory trials. That should change.
Two promising policy proposals are already advancing this idea. One comes from CASPR, which has proposed an “NIH High Leverage Trials Program”. In this program, a special body within NIH would prioritize and fund large-scale trials that could test new uses for off-patent medications and other interventions. Government support for large-scale trials is also part of the Cures in Care Initiative, part of Congressman Auchincloss’ bipartisan clinical trials framework. This initiative would have the government identify and potentially fund disease area-specific “platforms” that could execute large-scale trials embedded in routine care settings, including trials of new therapeutics. Both frameworks could create the impetus for NIH to reinvest in this important area.
But it’s not enough to just fund the trials themselves. NIH’s investments will go further if it commits to building reusable, national-level infrastructure to make trial execution easier for everyone. NIH should embrace interoperable trial technology; set standards for recurring trial operations like contracting and site qualification; and invest in reusable data infrastructure for trial matching, feasibility, monitoring, and follow-up. Then, after NIH-funded trials are completed, NIH should capture lessons learned and work with FDA to turn them into recognized trial practices that reduce operational burden and encourage innovation.
Crucially, this infrastructure investment is only possible if NIH serves as an important funder and operator of large-scale multi-center pivotal trials. Were this to take place, it would restore NIH to its historic role as a pioneer of innovation in late-phase trial design and conduct. In 2005, NIH conducted 230 phase 3 trials; by 2015 they funded only 62. It’s time to reverse that trend. NIH is the organization with both the scale and mandate to advance innovative trial designs, de-risk them, and build the infrastructure we need to execute them at scale.
Making efficient trials the default — not the exception
Together, these proposals would help efficiency-improving clinical trial innovations spread faster. An FDA program to recognize modern clinical trial practices can reduce regulatory uncertainty. Technology interoperability standards make it easier to develop and deploy innovative tools and technologies. And greater government investment in phase III trials can pressure-test innovative trial designs and support the development of trial infrastructure that others can reuse.
These policies alone won’t modernize trials. But they will help lay the groundwork on which other innovations can be built. Then, the work will fall on both the public and private sector to build and adopt meaningful innovations that improve trial efficiency, increase patient access, and create clinical trial abundance.
Here, I am optimistic. If we have a trial system that is capable of scaling up innovations, then the innovators themselves have the chance to step in and do what they do best. There are plenty of ideas waiting to be scaled up: new Bayesian and adaptive trial designs, AI-driven automation of trial operations, decentralized and point-of-care trials that allow patients to access trials in their community, and trials that leverage digital health technologies to gather more comprehensive data more cheaply. We know these kinds of innovations are possible. These policies can help make them the default.